



Comment identifier les gènes responsables de maladies héréditaires ?
C'est à partir des années 1990 que les généticiens ont cherché les gènes responsables de surdités héréditaires, par exemple en France dans le laboratoire du Professeur Christine Petit (Institut Pasteur, Paris). Depuis 1995, une centaine de gènes ont été identifiés. Comment identifier ces gènes ? En prenant en compte des familles où plusieurs individus, en particulier de plusieurs générations, sont atteints de surdité, d'une part, et en considérant des populations où la proportion de personnes atteintes de surdité est plus importante (on parle d'isolat géographique) que celle dans la population générale (environ 1 personne sur 1000 est atteinte de surdité héréditaire).
On peut séquencer le génome des individus appartenant à ces groupes (c'est-à-dire obtenir pour chaque gène l’enchaînement de nucléotides qui détermine la nature de la protéine obtenue à partir du gène) et comparer le génome des personnes atteintes de surdité à celui des personnes non atteintes. Cette comparaison permet d'identifier le gène atteint dans une famille ou une population données : ce gène se présente sous une version « saine » et une version « mutée ». La version « saine » du gène permet la fabrication d'une protéine qui joue un rôle donné dans des cellules données alors que la version « mutée » ne permet pas la fabrication de cette protéine ou alors celle d'une une protéine défectueuse. Cela a pour conséquence un dysfonctionnement des cellules ou cette protéine est normalement produite, ce qui aboutit ici à une surdité. Notons qu'il s'agit du schéma le plus simple, on peut en effet imaginer que plusieurs gènes soient atteints en même temps au lieu d'un seul.
Chaque individu a pour chaque gène deux versions : l'une provenant du père et l'autre de la mère (sauf pour ceux des chromosomes X et Y chez les hommes). Dans la plupart des cas (mais il y a de nombreuses exceptions) une personne naît sourde si elle a deux versions « mutées », c'est-à-dire que son père et sa mère la lui ont transmise. Dans ce cas, les parents ne sont pas nécessairement sourds (s'ils n'ont chacun qu'une version « mutée » et l'autre « saine »).
Comprendre le rôle des gènes responsables de maladies héréditaires
L'identification d'un gène responsable de maladie héréditaire, par exemple de surdité, ne donne toutefois pas la fonction de ce gène, l'explication de la surdité. Après que les généticiens ont identifié un gène responsable de surdité, le biologiste se demande : dans quelles cellules ce gène est-il « exprimé » (c'est-à-dire dans quelle cellule ce gène joue-t-il un rôle, où est produite la protéine correspondante) ? Quel rôle joue la protéine correspondante dans ces cellules ? Quelle est la conséquence de la mutation du gène sur le fonctionnement de ces cellules ?
Généralement, on répond à ces questions d'abord chez un animal proche de l'humain, par exemple la souris dont le système auditif ressemble à celui de l'humain. Dans le cas des gènes responsables de surdité, ils étaient tous « exprimés » dans l'oreille interne (un os en forme d'escargot rempli d'un liquide), situé après trois osselets et le tympan, la membrane que regarde l'ORL. On sent au touché cet os très dur (le plus dur de notre corps) à l'arrière de notre « oreille » ou plus précisément de notre oreille externe. Plus particulièrement, la plupart de ces gènes étaient exprimés dans les cellules sensorielles auditives. Ces cellules présentent des structures appelées microvillositées. Ces microvillosités détectent, en oscillant, des vibrations dans le liquide de l'oreille interne générées par les sons. C'est ainsi que l'oreille détecte les sons. Les cellules sensorielles auditives permettent ensuite la génération d'un signal électrique, transmis indirectement au cerveau via le nerf auditif.
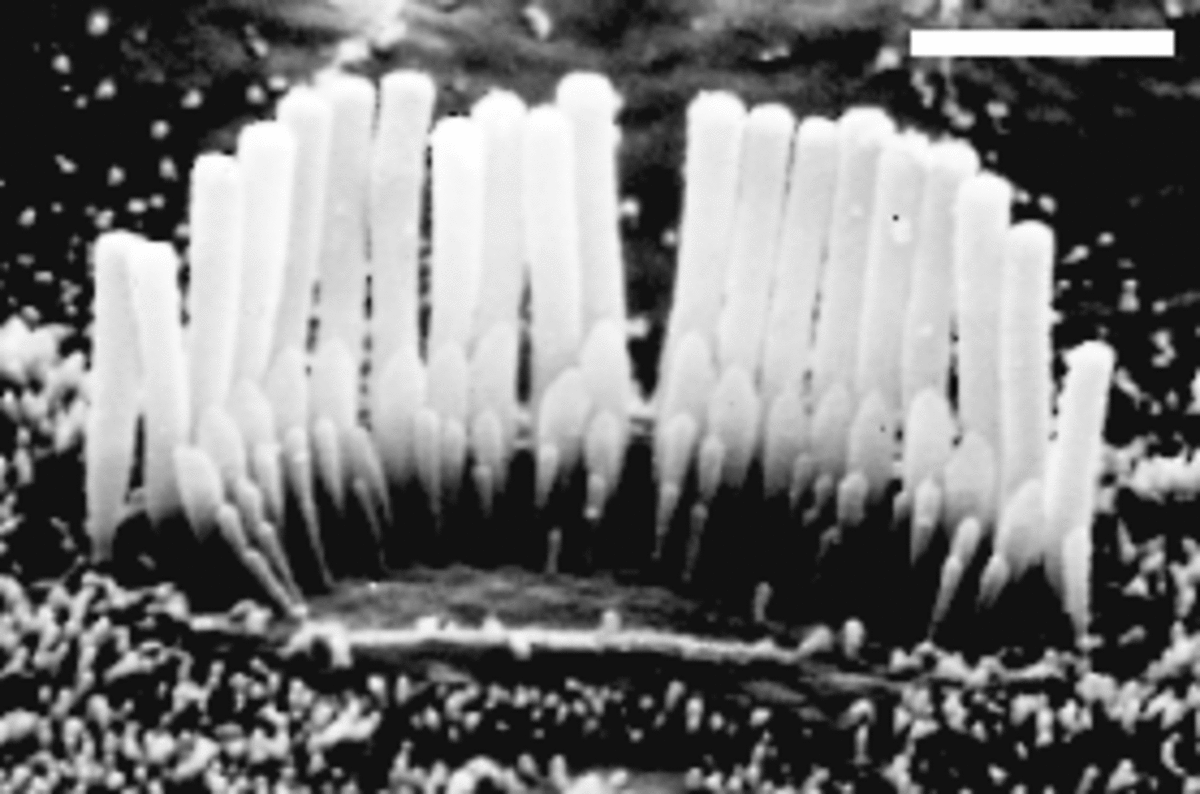
Nous avons les gènes, les cellules où ils sont exprimés, mais quel rôle joue les protéines associées à ces gènes ? Certaines d'entre elles forment des liens entre les microvillosités des cellules sensorielles auditives. Lorsque les microvillosités oscillent quand des vibrations se propagent dans l'oreille interne, elles tirent sur ces liens qui les relient entre elles. L'augmentation de la tension de ces liens permet l'ouverture à leur base de « canaux ioniques » qui permettent l'entrée de molécules chargées (des ions calciques par exemple) dans la cellules sensorielle. C'est cette entrée d'ions qui permet indirectement la formation d'un signal électrique qui sera transmis au cerveau. Les deux protéines qui forment ces liens entre les microvillosités des cellules sensorielles auditives sont appelées cadhérine-23 et protocadhérine-15. Les cadhérines sont des protéines qui permettent au cellules de « s'accrocher » les unes aux autres comme du scratch.
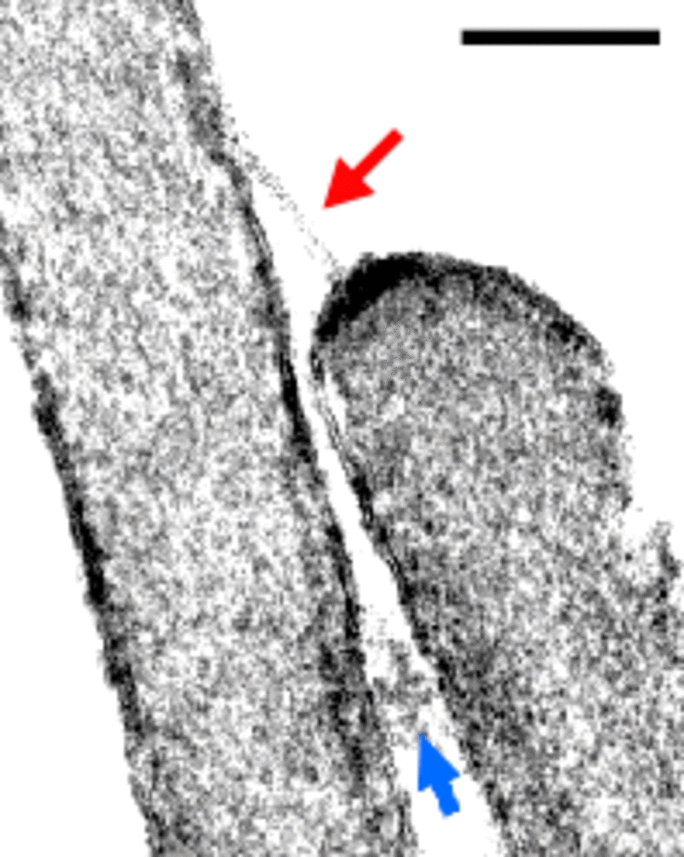
Afin d'avoir la preuve que c'est l'expression de ces gènes dans ces cellules sensorielles auditives qui était responsable de la surdité, des souris avec deux versions « mutées » de ces gènes ont été produites (pour obtenir la même configuration que les personnes atteintes de surdités). La biotechnologie a même permis d'avoir ces deux versions « mutées » uniquement dans les cellules sensorielles, et non dans le reste du corps. Cela a été suffisant pour que ces souris soient sourdes et présentent les mêmes signes de surdité que les patients chez qui ont avait identifié ces gènes.
Afin de permettre aux patients atteints de surdité profonde (ils ne perçoivent absolument aucun son), des implants cochléaires ont été créés. Ces implants remplacent les cellules sensorielles auditives : lorsqu'un son parvient à l'oreille, l'implant cochléaire génère un signal électrique transmis au cerveau.
Quid des régions cérébrales auditives chez les patients atteints de surdité ?
La pose des implants cochléaires a permis de manière remarquable aux patients atteints de surdité de percevoir des sons. La pose de ces implants part cependant de l'hypothèse que les gènes responsables de surdités ne sont « exprimés » que dans les cellules sensorielles auditives. Les régions auditives du cerveau ne seraient donc pas atteintes et pourraient jouer parfaitement leur rôle : par exemple, identifier d'où vient un son (ce qui est important dans la rue pour localiser d'où vient une voiture), ne faire attention qu'aux sons d'intérêt (dans une voiture on fait rapidement moins attention au bruit du moteur, par contre on fait attention à la radio ou aux personnes qui parlent), distinguer les différentes syllabes d'un mot, puis les mots dans une phrase, ou encore distinguer l'intonation (ce qui est important dans les langues comme le chinois, le vietnamien ou le thaï).
Or, mon travail de thèse, réalisé dans le laboratoire et sous la direction du Professeur Christine Petit (Institut Pasteur et Université Pierre-et-Marie-Curie, Paris), a permis d'illustrer le fait que certains gènes responsables de surdité, par exemple ceux associés à la cadhérine-23 et à la protocadhérine-15, qui forment les liens entre les microvillosités des cellules sensorielles auditives, jouent aussi un rôle dans la mise en place d'une région cérébrale auditive qu'on appelle le cortex cérébral auditif. Le cortex cérébral est situé à la surface de notre cerveau et sa région impliquée dans l'audition est située à peu près au dessus de notre oreille externe (même si l'oreille et le cortex auditif ne sont pas directement connectés entre eux). Le cortex cérébral auditif est impliqué dans la perception consciente des sons et aux tâches cognitives associées à l'audition (par exemple la compréhension du langage).
Nous avons en effet montré chez la souris et chez le macaque que ces deux protéines sont « exprimées » au cours du développement embryonnaire du cortex cérébral auditif (de manière équivalente chez l'humain, du 3ème au 6ème mois de grossesse). Plus particulièrement elles sont « exprimées » dans des futurs neurones qu'on appelle « neurones inhibiteurs ». Ces neurones régulent l'activité cérébrale, notamment en inhibant l'activité des neurones qui sont à leur proximité. Au cours du développement embryonnaire, les futurs « neurones inhibiteurs » naissent au cœur du cerveau puis migrent (à une vitesse de 2 mm par jour) vers leur destination finale. Ces futurs neurones ont besoin de différentes protéines pour pouvoir avancer, et se diriger dans la bonne direction. Nous avons montré que la cadhérine-23 et la protocadhérine-15 permettent à certains futurs « neurones inhibiteurs » de se diriger uniquement vers le cortex cérébral auditif. Elles agissent en quelque sorte comme des codes-barres qui permettent aux futurs neurones d'être « adressés » à un endroit donné. C'est le premier exemple d'un tel « adressage » de « neurones inhibiteurs ».
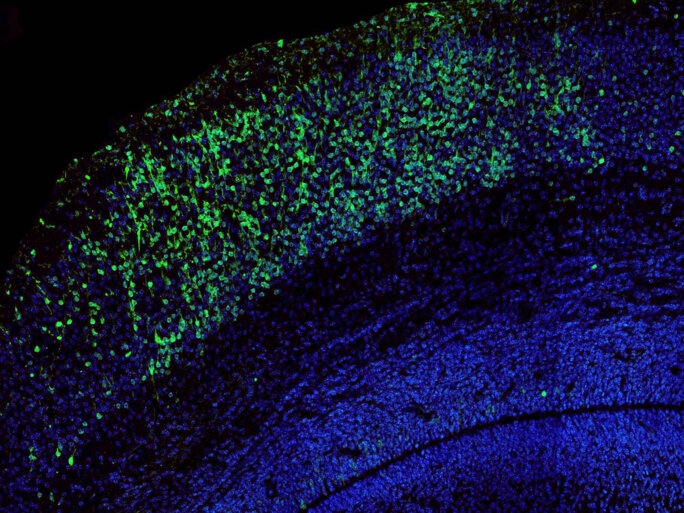
Agrandissement : Illustration 3
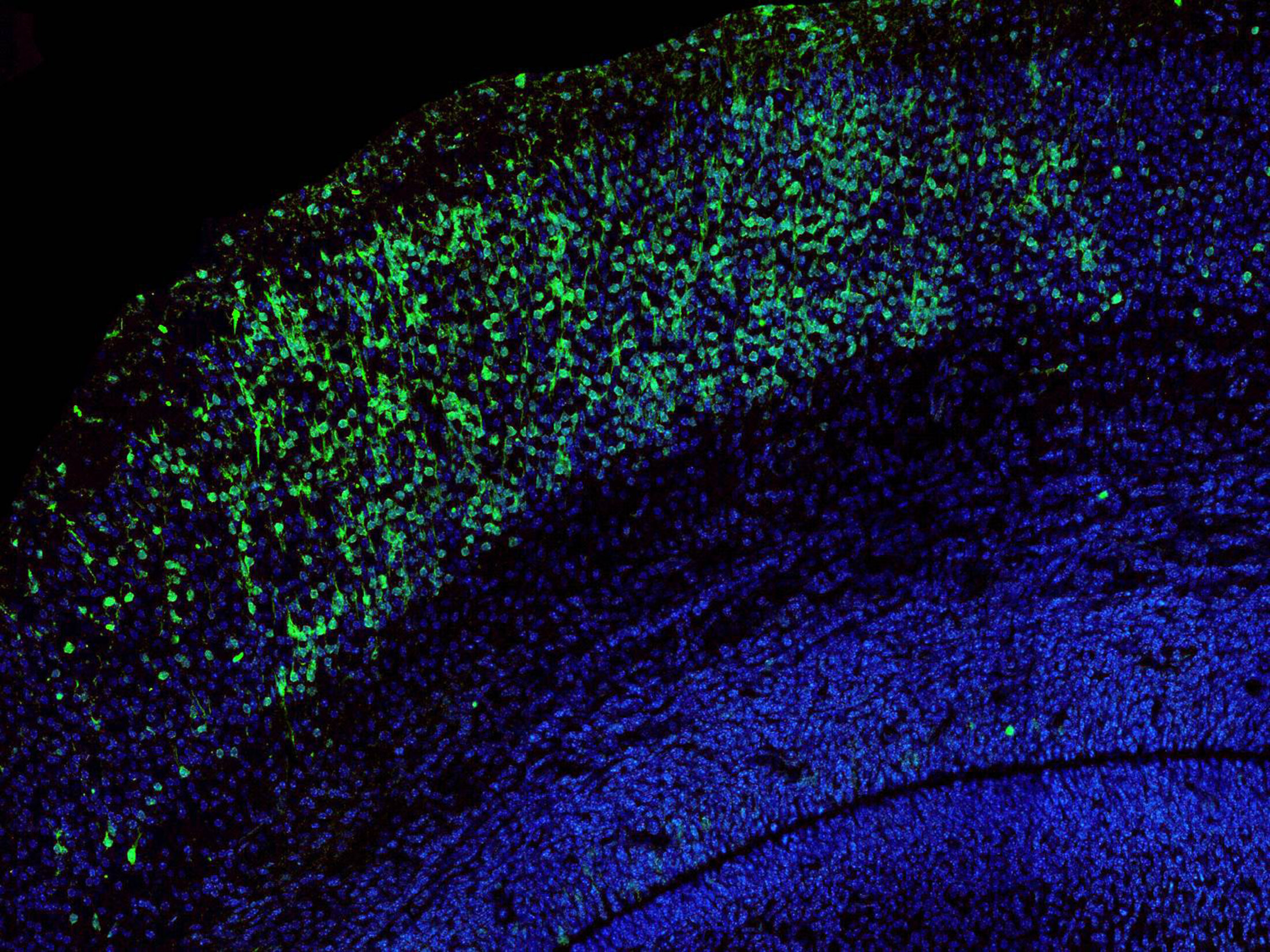
Lorsque des souris ont deux versions « mutées » des gènes associés à la cadhérine-23 et à la protocadhérine-15, voire deux versions « mutées » uniquement dans les futurs « neurones inhibiteurs », ces neurones ne parviennent pas à atteindre le cortex cérébral auditif. Ces souris présentent dès leur naissance moins de « neurones inhibiteurs » dans le cortex cérébral auditif, qui est ainsi « dérégulé".
Il faudra vérifier que cette anomalie du cortex cérébral auditif est aussi présente chez les patients avec des versions « mutées » pour les gènes associés à la cadhérine-23 et la protocadhérine-15, même si leur « expression » dans les futurs « neurones inhibiteurs » du cortex cérébral auditif chez le macaque le laisse penser. De plus, il faudra préciser de quelle manière le cortex cérébral auditif est « dérégulé » chez ces patients. On peut s'attendre par exemple à des défauts de compréhension du langage, par exemple distinguer des phonèmes successifs. Les « neurones inhibiteurs » atteints sont aussi impliqués dans ce qu'on appelle la « plasticité cérébrale », permettant au cortex cérébral auditif de maturer en fonction de son environnement sonore. Cela devra être pris en compte dans l'accompagnement des patients, souvent des enfants, suite à la pose d'un implant cochléaire.
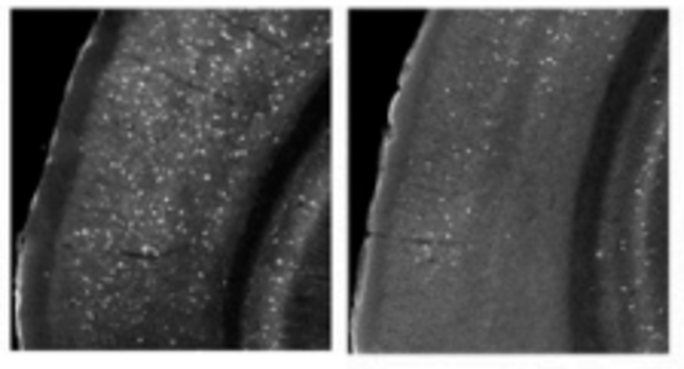
Nos résultats illustrent aussi que le cortex cérébral auditif peut être atteint indépendamment de l'oreille interne. De nombreuses personnes présentent des troubles auditifs (mauvaise audition dans le bruit, mauvaise compréhension du langage), sans qu'aucun défaut ne soit détecté par l'ORL (testant le fonctionnement de l'oreille interne). Des tests devront donc être élaborés pour tester le fonctionnement de leur cortex cérébral auditif.
Notes :
Ont participé à cette étude : Baptiste Libé-Philippot, Vincent Michel, Jacques Boutet de Monvel, Sébastien Le Gal, Typhaine Dupont, Nicolas Michalski et Christine Petit (Institut Pasteur/Inserm/Université Pierre-et-Marie-Curie/Collège de France, Paris), Christine Métin (Institut du Fer à Moulin/Inserm/Université Pierre-et-Marie-Curie, Paris) et Paul Avan (Université d'Auvergne/Inserm, Clermont-Ferrand). Cette étude a été rendue possible par des financements issus de l'Agence nationale pour la recherche (ANR), de la Commission européenne (European Research Council), du Ministère de l'Enseignement supérieur et de la Recherche (allocation doctorale) et de la fondation Agir pour l'audition.
