



spectrumnews.org Traduction de "Fragile X syndrome traits may stem from leaky mitochondria | Spectrum | Autism Research News" par Angie Voyles Askham / 11 septembre 2020
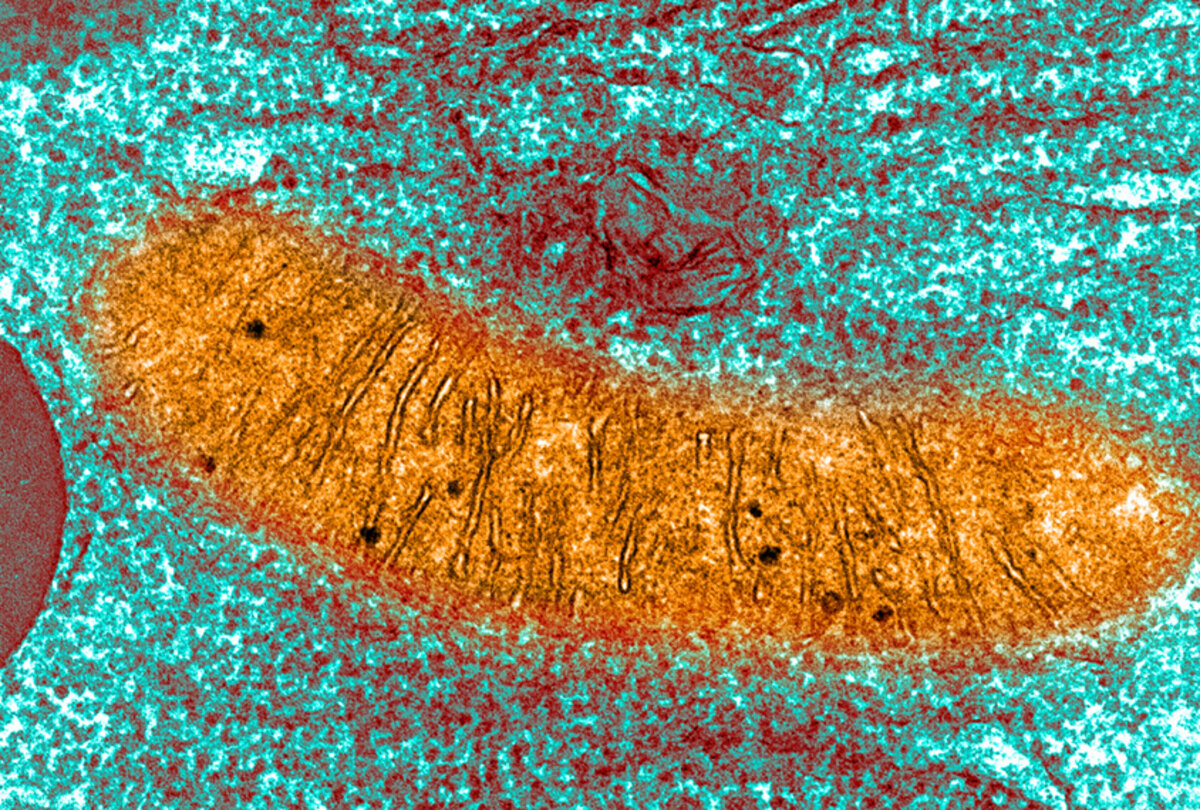
Agrandissement : Illustration 1
De nombreux problèmes associés au syndrome X fragile proviennent d'une fuite dans les mitochondries, des organites qui agissent comme les centrales électriques de la cellule, suggère une nouvelle étude 1. En stoppant cette fuite, on atténue certains des traits autistiques des souris qui reproduisent le syndrome.
"Le papier est très solide", affirme John Jay Gargus, directeur du Centre de recherche et de traduction sur l'autisme de l'Université de Californie à Irvine, qui n'a pas participé à l'étude. Et comme le déficit en énergie mitochondriale est observé dans d'autres formes d'autisme, les conclusions pourraient être pertinentes au-delà du syndrome X fragile, selon M. Gargus.
Le syndrome X fragile résulte de mutations dans le gène FMR1, qui entraînent une perte de la protéine FMRP. Sans FMRP, les cellules ont des épines dendritiques immatures - les bosses le long des bras d'un neurone qui reçoivent l'apport d'autres neurones - et produisent d'autres protéines en excès. On pense que ces différences contribuent aux traits caractéristiques du syndrome, tels que le retard de développement, la déficience intellectuelle et, souvent, l'autisme.
La nouvelle étude montre qu'une lésion de la membrane mitochondriale, probablement causée par l'absence de FMRP, peut entraîner l'immaturité des cellules touchées et une production excessive de protéines. La lésion affecte le métabolisme d'une cellule, l'obligeant à produire de l'énergie rapidement mais pas efficacement, explique la chercheuse principale Elizabeth Jonas, professeure de médecine interne et de neuroscience à l'université de Yale.
Toutes les cellules commencent par des fuites mitochondriales ; la production rapide d'énergie que permettent ces fuites peut être utile au début du développement. Cependant, à mesure que les cellules typiques mûrissent et que l'efficacité devient plus importante que la vitesse, elles semblent colmater les fuites, explique Mme Jonas. Comme les cellules présentant une mutation X fragile ne peuvent pas fermer leurs brèches, elles restent dans un état immature.
L'application d'un composé qui bloque les brèches conduit à un métabolisme plus mature et diminue la production de protéines dans les neurones de souris modèles du syndrome X fragile et dans les fibroblastes de personnes atteintes du syndrome, ont constaté les chercheurs. Et l'injection de ce composé à des souris modèles du syndrome X fragile diminue leurs comportements de type autiste.
"Cela montre une très belle cible : un canal unique qui peut être manipulé", explique le chercheur Pawel Licznerski, chercheur associé en médecine à l'université de Yale.
Des membranes qui présentent des fuites
Jonas, Licznerski et leurs collègues ont observé que les mitochondries des souris X fragiles sont plus petites et produisent moins d'adénosine triphosphate (ATP), la molécule qui transfère l'énergie à travers les cellules, par rapport aux témoins.
Une enzyme en forme de sucette intégrée dans la membrane mitochondriale, appelée ATP synthase, produit de l'ATP en pompant les ions hydrogène dans la chambre interne de l'organelle et en les empêchant de s'échapper.
Pour voir si la pénurie d'ATP était due à un problème d'ATP synthase, les chercheurs ont isolé des vésicules - des bulles de membrane mitochondriale contenant de l'ATP synthase - de souris X fragiles et de souris témoins. Ils ont placé les vésicules dans un bain contenant un colorant qui devient fluorescent en présence d'ions hydrogène.
L'ATP synthase des souris typiques a pompé tous les ions hydrogène dans les vésicules et les a piégés, diminuant l'intensité de la fluorescence dans le bain. Mais le bain contenant les vésicules de souris X fragile est resté très fluorescent ; les vésicules X fragile "ne pouvaient pas séquestrer d'ions hydrogène", explique Jonas.
Les vésicules des souris modèles de l'X fragile avaient également une "conductance membranaire" nettement plus élevée, signe que les ions hydrogène entrent et sortent continuellement des vésicules. Ce résultat suggère que la synthase peut pomper les ions à l'intérieur, mais qu'ils s'échappent d'une manière ou d'une autre à travers la membrane.
Les chercheurs ont découvert que le traitement des vésicules avec le composé dexpramipexole bloquait la fuite et diminuait la conductance. Les travaux ont été publiés en août dans Cell.
Boosts métaboliques
L'ATP synthase comprend de multiples sous-unités. Par rapport aux mitochondries des neurones typiques, celles des neurones X fragiles comportent une proportion nettement plus importante d'une sous-unité, appelée sous-unité C, ont constaté les chercheurs. Ce déséquilibre pourrait expliquer pourquoi ces mitochondries ne peuvent pas piéger les ions hydrogène : Les sous-unités c peuvent exister par elles-mêmes en tant que canaux dans la membrane mitochondriale et permettre aux ions hydrogène de s'échapper.
Pour fermer ces canaux, une cellule doit incorporer les sous-unités c dans de nouvelles synthases d'ATP, un processus qui nécessite une autre sous-unité, appelée bêta. Les chercheurs ont découvert que la protéine manquante chez les personnes atteintes du syndrome X fragile, la FMRP, régule la fabrication des sous-unités bêta. Incapables de réguler la façon dont les sous-unités bêta sont produites, Jonas et ses collègues affirment que les mitochondries des cellules des souris atteintes du syndrome X fragile restent perméables.
La perméabilité persistante influence la voie métabolique que la cellule utilise pour produire de l'énergie, a découvert l'équipe en utilisant une technique appelée spectrométrie de masse.
Par exemple, les neurones X fragiles produisent plus d'enzymes associées à la glycolyse - une voie couramment utilisée par les cellules immatures - que les neurones typiques. Des études antérieures ont montré une altération du métabolisme mitochondrial chez les personnes atteintes d'autres formes d'autisme 2.
L'ajout de dexpramipexole aux cellules de souris X fragiles a diminué la production de lactate déshydrogénase et d'autres enzymes liées à la glycolyse, ce qui suggère que la fermeture de la brèche fait que les neurones commencent à utiliser des voies métaboliques différentes et plus matures.
L'injection de dexpramipexole à des souris modèles de l'X fragile a réduit leur hyperactivité, leurs comportements répétitifs et leur toilettage excessif - des traits qui rappellent ceux observés chez les personnes autistes et chez celles atteintes du syndrome X fragile. Les souris qui ont reçu les injections de dexpramipexole avaient également des neurones avec des épines dendritiques plus matures et des niveaux réduits de synthèse de protéines.
Changements de canaux
Bien qu'intéressantes, les conclusions "ne fournissent pas de mécanisme clair sur la façon dont la FMRP régule la fuite", explique Xinyu Zhao, professeur de neuroscience à l'université du Wisconsin-Madison, qui n'a pas participé à l'étude. Les recherches de Zhao et de ses collègues suggèrent qu'une interaction entre la FMRP et une autre protéine est nécessaire pour que les mitochondries fonctionnent correctement et que les cellules mûrissent 3.
Même sans mécanisme, le fait de pouvoir atténuer les traits caractéristiques de l'autisme chez les souris en fermant un canal membranaire "est remarquable", selon Licznerski. "Cela ouvre la porte à une nouvelle approche de la thérapie" pour les personnes atteintes du syndrome de l'X fragile, dit-il.
D'autres recommandent la prudence : Il n'y a aucune garantie qu'un traitement qui fonctionne chez les souris fonctionnera dans le cerveau humain, déclare Cecilia Giulivi, professeur de biosciences moléculaires à l'université de Californie, Davis MIND Institute, qui n'a pas participé à l'étude. Elle craint que ces résultats préliminaires n'offrent de faux espoirs aux familles des personnes atteintes du syndrome X fragile et n'encouragent les gens à rechercher des traitements dont l'innocuité n'a pas été prouvée. Le dexpramipexole a été testé chez des personnes atteintes de la maladie neurologique de la sclérose latérale amyotrophique et s'est révélé sûr, mais on ne sait pas exactement comment il affecterait les jeunes s'il était pris sur des périodes prolongées.
Avant qu'un traitement soit possible, Jonas et Licznerski disent qu'ils doivent mieux comprendre comment les sous-unités de l'ATP synthase contribuent à la fuite. Dans ce but, ils étudient des souris sans sous-unités c supplémentaires pour voir si le fait de minimiser la fuite de cette manière a le même effet que l'application de dexpramipexole.
Références: