



Tandis que le variant Omicron devient peu à peu majoritaire dans le cadre de nouvelles infections1, que la barre des 300 000 contaminations quotidiennes en France s’approche, une des conséquences directes concerne l’explosion du nombre de tests de dépistage dit « PCR ». Face à l’évolution du virus menant à de nouveaux « variants » (Alpha, Bêta, Gamma, Delta, Omicron et autres lettres de l’ancien alphabet grec), comment se comportent et évoluent les techniques de détection ? Pour comprendre, il est déjà nécessaire de savoir de quoi l’on parle… Et de ce fait, c’est quoi une PCR ?
La PCR : principe d’une méthode fiable qui a fait ses preuves depuis longtemps
La fameuse PCR, ou Réaction de Polymérisation en Chaîne (Polymerase Chain Reaction pour les anglophones), est une technique de biologie moléculaire qui est née à la fin des années 19802 et qui fait désormais loi dans de nombreux laboratoires de recherche en génétique. Je me souviens d'un professeur qui m'avait expliqué jadis que "la PCR est au biologiste ce que le tricycle est à l'enfant !". Mais en gros, en quoi consiste-t-elle ? Et pourquoi est-elle aussi utilisée ? Le tout est question de précision et de fiabilité…
L’idée est de détecter dans un échantillon biologique (tissu, crachas, sang, etc.) une séquence d’ADN spécifique (ça marche aussi pour l’ARN, mais je vais y revenir), et d’amplifier sa présence afin de pouvoir déterminer sa présence, ou à défaut d’amplification, son absence. C’est un peu comme chercher une aiguille dans une botte de foin, mais en mode « tricherie » : est-ce que cette botte de foin contient l’aiguille que je cherche ? Rajoutons un produit magique à la botte de foin pour démultiplier spécifiquement l’aiguille que je cherche (entre 235 et 240 fois, c’est-à-dire entre environ 35 milliards et 1 100 milliard de fois), et uniquement cette aiguille précise. Si elle est bien présente, je serai capable de la voir au final, car je l’aurai multiplié des milliards de fois… Si elle est absente, il n’y aura pas d’aiguille visible.
Comment une telle multiplication peut-elle être aussi précise ? Tout vient de son processus, de la recette de cuisine appliquée… Pour qu’une telle multiplication se fasse dans notre échantillon, il nous faut un effecteur, une molécule qui va être capable de se fixer sur l’ADN de l’échantillon, et de multiplier la zone ciblée. Cet effecteur s’appelle une ADN polymérase2, une enzyme (c’est-à-dire un catalyseur, un accélérateur de réaction) qui peut se fixer (on parle du terme « s’hybrider ») sur une zone précise de l’ADN, pour amplifier/multiplier cette zone. Mais comment cette molécule peut-elle se fixer à un endroit précis de l’ADN, et pas à un autre ? Eh bien celle-ci va être guidée/orientée par ce que l’on appelle des amorces ADN (ou primers anglais).
Ces amorces sont des séquences d’ADN courtes, typiquement d’une longueur de 20 à 25 nucléotides (les briques constitutives de l’ADN), qui ont pour propriété d’être complémentaires à l’ADN ciblé. Car oui, l’ADN est composé de 4 types de briques : A, C G et T. Il s’avère que ces briques présentent une complémentarité particulière : le A et le T s’attire, tout comme le C et le G. Ainsi, si on prend l’exemple d’une séquence d’ADN qui ressemblerait à ATTAGCCATA, celle-ci va être complémentaire à la séquence TAATCGGTAT (voir la figure associée).
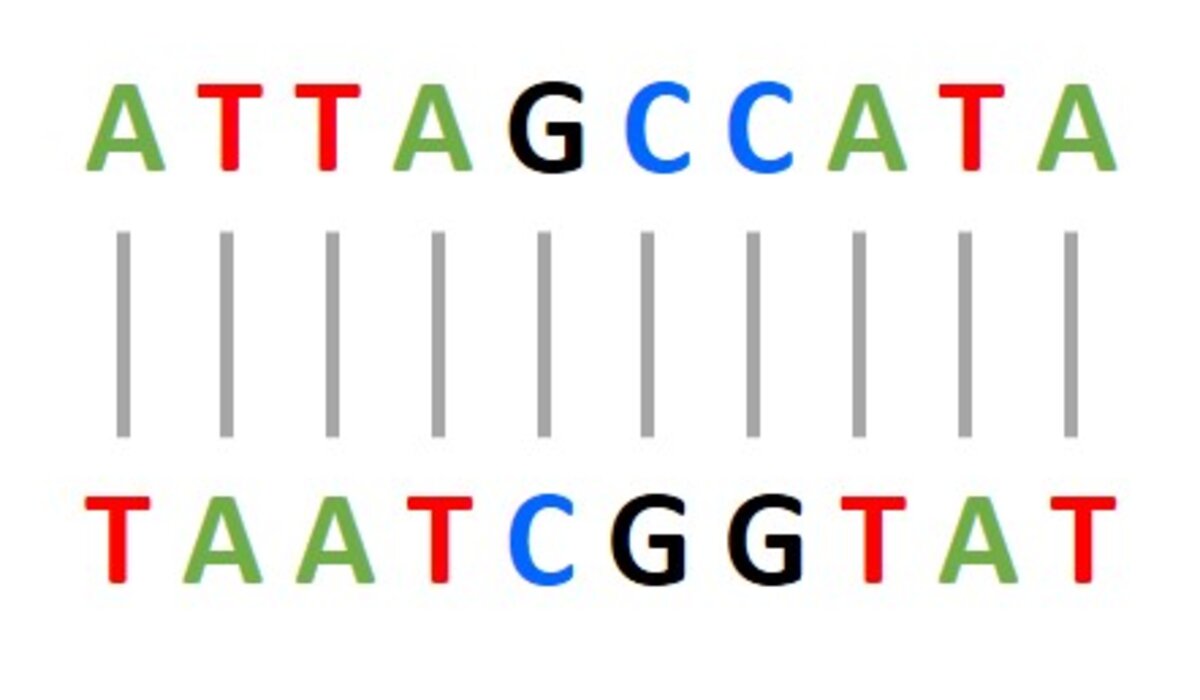
On peut se dire que 20 briques, c’est court, mais la probabilité d’avoir une séquence particulière de 20 briques par hasard est de 420, soit 1 chance sur 1 099 511 627 776 (1 chance sur 1 100 milliards). Ce qui laisse finalement bien peu de place au hasard ! Bien sûr, la réalité est plus complexe, car certaines séquences d’ADN sont répétées, et existent donc en plusieurs copies au sein d’un même organisme, mais le principe de fiabilité est bien là… Ainsi, une PCR va amplifier l’ADN encadré par un couple d'amorces (spécifiques de la zone ciblée) via l’ADN polymérase… A partir de là, la PCR va consister en différents cycles de variation de température pour maximiser l’efficacité de la réaction : à chaque cycle, l’ADN ciblé verra sa quantité multipliée par 2, ainsi une PCR de 35 cycles va potentiellement amplifier l’ADN cible 235 fois, de 40 cycles 240 fois, etc. D’où les ordres de grandeur cités précédemment.
La PCR et ses dérivés… C’est quoi, une RT-PCR quantitative ?
A partir de cette technique, de nombreuses déclinaisons ont pu être développées… Comme nous le savons tous désormais, le SARS-CoV2 est un virus à ARN3–5 et non à ADN. Ainsi soit-il, il est possible pour les biologistes de « convertir » de l’ARN en ADN, qui est bien plus maniable et manipulable (car plus stable) que l’ARN. On parle de réaction de Réverse-Transcription, ou de RétroTranscription, qualifiée sous l’abréviation RT. L’étape de RétroTranscription permet de générer de l’ADN complémentaire (ADNc) à partir d’ARN… Donc on peut lever un premier abus de langage, lorsque l’on fait un test de dépistage PCR, en réalité c’est une réaction de RT-PCR qui est réalisée ! D’autant plus que les tests de dépistage donnent une réponse un peu plus présence qu’une « simple » présence (amplification) ou absence (pas d’amplification)…
En effet, un autre dérivé de la PCR permet même de quantifier l’ADN (ou l’ADNc) source ayant servi de base à la réaction ! Plus il y a d’ADN ciblé au départ dans notre échantillon, plus l’amplification sera rapide… Imaginons un exemple fictif : (i) suite à un prélèvement nasal, un seul virus SARS-CoV2 est présent dans l’échantillon. Après 35 cycles de PCR, il y aura 235 copies de la séquence virale ciblée. (ii) A présent, suite à un autre prélèvement, 1 000 virus sont présents… Eh bien après 35 cycles de PCR, nous aurons 1 000 x 235 copies de la séquence virale ciblée ! Pour ainsi atteindre le même nombre de copies amplifiées dans les 2 cas, le cas (ii) ira donc bien plus vite que le cas (i). C’est là le principe de la PCR quantitative, communément appelée PCR en temps réel ou encore qPCR. Dans le cas du SARS-CoV2, la méthode de détection employée est une combinaison des approches de RT-PCR et de qPCR, à savoir une technique de RT-qPCR !
Evolution du virus et spécificité des amorces
Si vous avez tout suivi jusque-là, les biologistes utilisent donc une technique assez complexe, particulièrement fine et précise pour la détection de notre « cher » virus… Mais l’ARN de notre virus n’est pas figé et statique dans le temps ! Sinon il n’y aurait pas les fameux variants dont on parle sans cesse… Des études dites « phylodynamique », qui correspondent d’une certaine façon à des études généalogiques, suggèrent que le taux de mutation du SRAS-CoV2 se situe entre 1,05 × 10-3 et 1,26 × 10-3 substitutions par site par an. Ce qu’il faut comprendre derrières ces termes à la limite de l’ésotérisme, c’est que comme tous les Coronaviridae6–8 (famille du SARS-CoV2), le virus mute rapidement et donc, évolue vite. Vous voyez donc peut-être là où je veux vous emmener… En effet, face à un virus qui évolue rapidement, il est nécessaire d’adapter les stratégies de détections d’autant plus que ces dernières ont pour principale faiblesse (qui est le plus souvent une force !) d’être extrêmement spécifique. Si le virus mute trop, les amorces ne peuvent plus se fixer sur la zone cible, et de facto l’amplification PCR n’aura pas lieu, alors que le virus est bien présent !
Une étude publiée en Avril 2021 dans le journal Scientific Reports9 a permis de recenser les protocoles utilisés par différentes localités (États-Unis, Chine, Allemagne, Hong-Kong, Thaïlande, France et Japon) afin d’évaluer la fiabilité de ses protocole au cours du temps. L’idée était de savoir à quel point les amorces utilisées étaient fiables, et à quelle vitesse évoluait leur fiabilité au fur et à mesure de l’évolution du virus lui-même. Ainsi, les chercheurs ont comparé ces amorces avec les différents génomes du SARS-CoV2 publiés au cours du temps sur une période de 207 jours (du 1er Janvier 2020 au 25 Juillet 2020), le tout couplé à diverses analyses spatio-temporelles (dont je passerai totalement les détails techniques ici).
Si on compare les amorces utilisées par différents pays avec les génomes du virus alors disponibles (on parle de 61 996 génomes utilisés dans la publication en question…), on constate que le nombre de mésappariements (mismatches), donc d’erreurs, entre les amorces et les génomes disponibles est fortement variable d’un protocole à l’autre, d’un pays à l’autre.
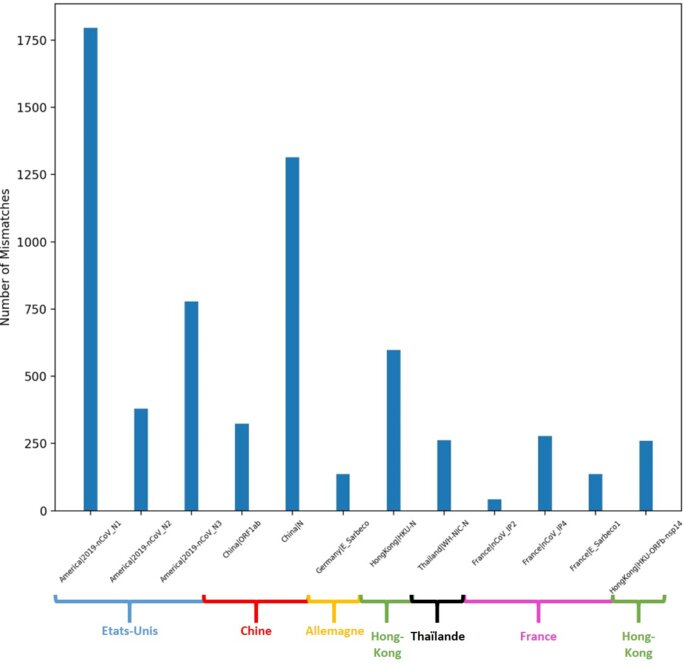
Agrandissement : Illustration 2
Mais pourquoi de telles différences selon les pays, et donc selon les protocoles utilisés ? Tout est à voir au niveau de la zone amplifiée par les amorces utilisées… Car toutes les zones du génome viral n’évoluent pas à la même vitesse ! Certaines sont en effet plus stables que d’autres, et les protocoles des différents pays ne ciblent pas nécessairement la même région du génome viral. Les amorces utilisées par les Etats-Unis, la Chine, Hong-Kong et la Thaïlande ciblant un gène codant pour une nucléo-protéine (en vert) semblent présenter un plus grand nombre de mésappariements… Pourquoi cibler une telle région ? Car elle est de très grande taille, donc facile à cibler10.
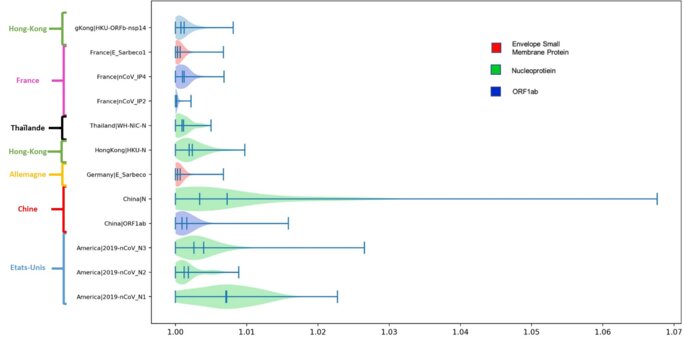
Agrandissement : Illustration 3

Néanmoins, les auteurs de l’étude suggèrent qu’il serait peut-être intéressant de dessiner de nouvelles amorces, ciblant des régions plus stables et moins sujettes aux mutations…
Avec l’émergence de mutations spécifiques qui se propagent à un rythme plus rapide (donnant naissance typiquement aux fameux « variants »), cette étude souligne et évalue la nécessité de revoir régulièrement la conception des amorces PCR utilisées, d’adapter notre paire de jumelles de détection face à virus qui peut changer au cours du temps…
Décidément, ce virus n’est VRAIMENT pas simple à détecter…
Références bibliographiques :
- Ferré, V. M. et al. Omicron SARS-CoV-2 variant: What we know and what we don’t. Anaesthesia, critical care & pain medicine 41, 100998 (2022).
- Saiki, R. K. et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487–491 (1988).
- Zhang, Y.-Z. & Holmes, E. C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 181, 223–227 (2020).
- Kim, D. et al. The Architecture of SARS-CoV-2 Transcriptome. Cell 181, 914-921.e10 (2020).
- Brant, A. C., Tian, W., Majerciak, V., Yang, W. & Zheng, Z.-M. SARS-CoV-2: from its discovery to genome structure, transcription, and replication. Cell & Bioscience 11, 136 (2021).
- Baric, R. S., Yount, B., Hensley, L., Peel, S. A. & Chen, W. Episodic evolution mediates interspecies transfer of a murine coronavirus. J Virol 71, 1946–1955 (1997).
- Cotten, M. et al. Spread, Circulation, and Evolution of the Middle East Respiratory Syndrome Coronavirus. mBio (2014) doi:10.1128/mBio.01062-13.
- Dudas, G., Carvalho, L. M., Rambaut, A. & Bedford, T. MERS-CoV spillover at the camel-human interface. eLife 7, e31257 (2018).
- Nayar, G. et al. Analysis and forecasting of global real time RT-PCR primers and probes for SARS-CoV-2. Sci Rep 11, 8988 (2021).
- Carter, L. J. et al. Assay Techniques and Test Development for COVID-19 Diagnosis. ACS Cent Sci 6, 591–605 (2020).